Haupt- und Nebenkriterien
bei Ehlers-Danlos-Syndromen

|
EDS Subtyp |
Prävalenz* |
Abkürzung |
Vererbung |
Assoziertes Gen |
Protein |
|
Klassisches EDS |
1 / 20 000 – 40 000 |
cEDS |
AD |
Major: COL5A1, COL5A1 Rare: COL1A1 c.934>T,p. (Arg312Cys) |
Kollagen Typ V Kollagen Typ I |
|
Hauptkriterien |
Nebenkriterien |
|
1. Überdehnbarkeit der Haut1 und atrophe Narbenbildung2 |
1. Schnelle Hämatombildung4 |
|
2. Generalisierte Gelenkhypermobilität (GJH)3 |
2. Weiche, teigige Haut5 |
|
3. Hautfragilität (oder traumatische Risse) |
|
|
4. Molluscoide Pseudotumoren6 |
|
|
5. Subkutane Sphäroide7 |
|
|
6. Hernie (oder Hernie(n) in der Vergangenheit) |
|
|
7. Epikanthusfalten8 |
|
|
8. Komplikationen der Gelenkhypermobilität (z. B. Verstauchungen, Luxation/Subluxation, Schmerzen, flexibler Plattfuß) |
|
|
9. Familienanamnese eines Verwandten ersten Grades, der die klinischen Kriterien erfüllt |
Minimalkriterien, die auf cEDS hinweisen:
- Hauptkriterium (1): Überdehnbarkeit der Haut und atrophe Narbenbildung
Plus
- Entweder Hauptkriterium (2): Generalisierte Gelenkhypermobilität
- Und/oder: mindestens drei Nebenkriterien
Die Bestätigung durch einen molekulargenetischen Tests ist erforderlich, um eine endgültige Diagnose zu stellen.
Fußnoten
* Prävalenzwerte stammen von der EDS Society
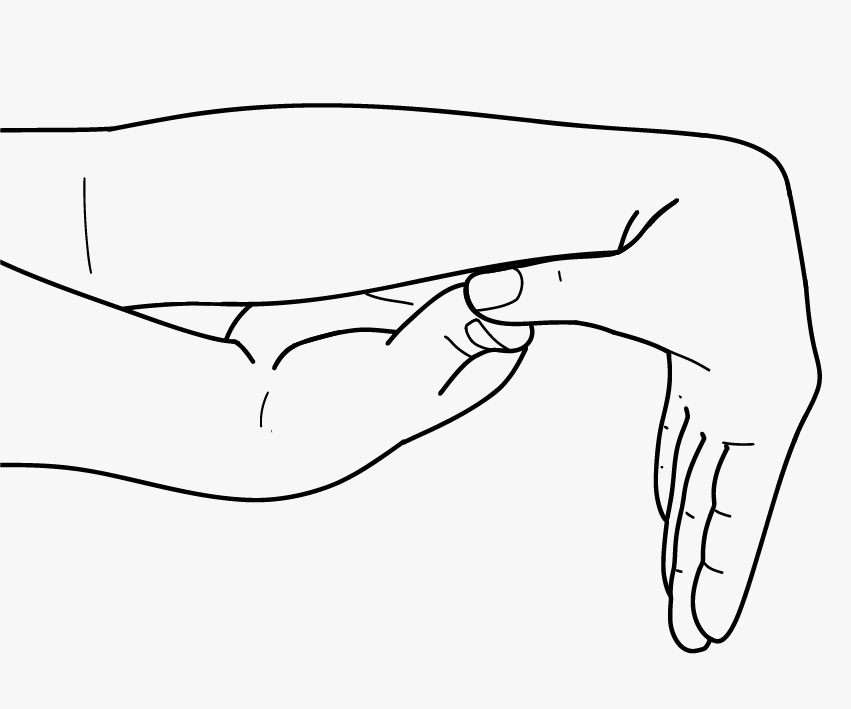
- Skin extensibility should be measured by pinching and lifting the cutaneous and subcutaneous layers of the skin on the volar surface at the middle of the non-dominant forearm as described in Remvig et al. [2009]. Skin is hyperextensible if it can be stretched over a standardized cut-off in three of the following areas: 1.5 cm for the distal part of the forearms and the dorsum of the hands; 3 cm for neck, elbow, and knees.
- Abnormal scarring can range in severity. Most patients have extensive atrophic scars at a number of sites (Fig. 1). These can sometimes be haemosiderotic. A minority of patients are more mildly affected.
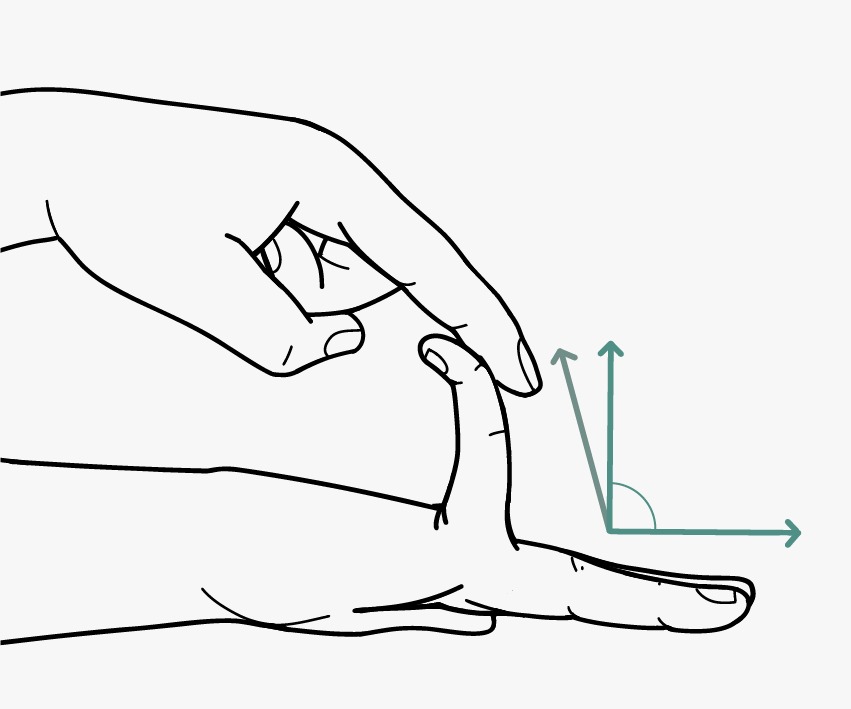
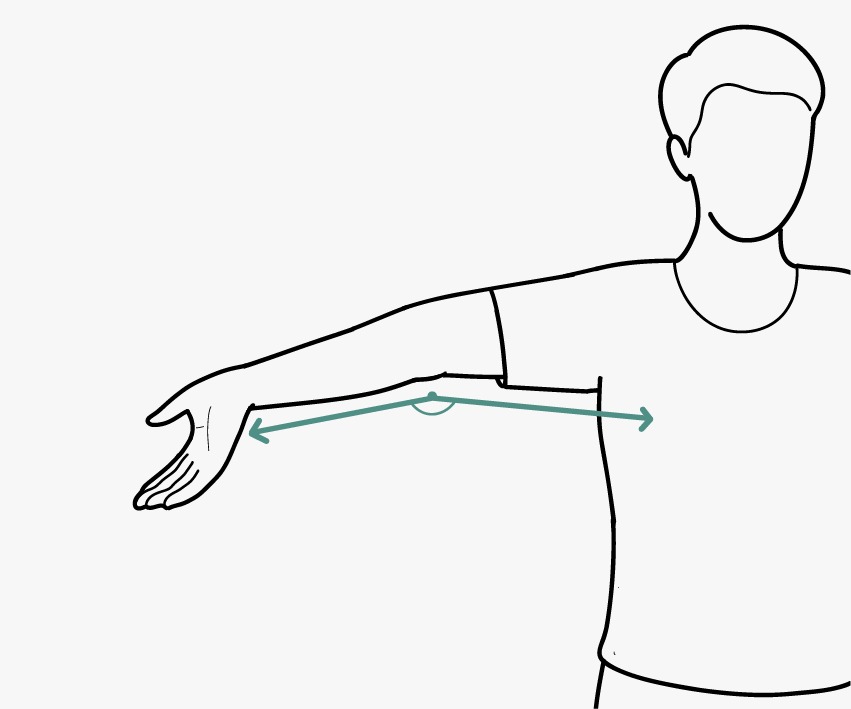
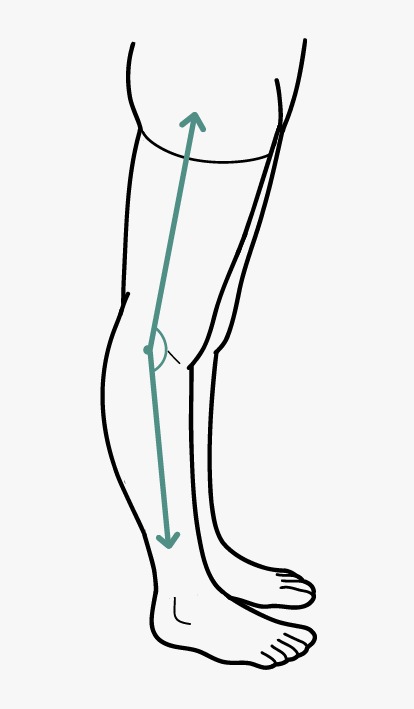
- GJH is evaluated according to the Beighton score; a Beighton score of ≥5 is considered positive for the presence of GJH (Fig. 2). Since laxity decreases with age, patients with a Beighton score <5/9 may be considered positive based on their historical observations (see “five-point questionnaire (5PQ)” (Table III).
- Easy bruising can occur anywhere on the body, including unusual sites. The pretibial area often remains stained with hemosiderin from previous bruises.
- Subjective abnormality of the skin texture is appreciable by touching the skin.
- Molluscoid pseudotumors are fleshy lesions associated with scars, found over pressure points (e.g., elbow, fingers).
- Subcutaneous spheroids (Fig. 1E) are small spherical hard bodies, frequently mobile, and palpable on the forearms and shins. Spheroids may be calcified and detectable radiologically.
- Epicanthal folds are often seen in childhood but may also be seen in adults.
- For definitions of GJH and skin hyperextensibility, see criteria for “Classical EDS.”
- The cardiac-valvular problems were reported in all affected adult individuals, but were absent in the two reported children (both <10 years of age).
- For definition of skin hyperextensibility, see criteria for “Classical EDS.”
- If marfanoid features are present, consider other conditions such as: Marfan syndrome, Loeys–Dietz syndrome, congenital contractural arachnodactyly, Shprintzen–Goldberg syndrome, Stickler syndrome, Homocystinuria, multiple endocrine neoplasia type 2B, and the familial thoracic aortic aneurysmal disorders [Pyeritz and Loeys, 2012]. Molecular testing for many of these conditions is clinically available.
- While skin softness and texture remain subjective, it is often very notable in some individuals and useful when present but not quantifiable; we therefore recommend a high threshold for positivity.
- Skin extensibility as measured by pinching and lifting the cutaneous and subcutaneous layers of the skin on the volar surface at the middle of the non-dominant forearm as described in Remvig et al. [2009]. Skin extensibility of >1.5 cm is considered the upper end of normal. It is likely that the hyperextensibility of the skin in hEDS overlaps significantly with that of “normal” skin. Therefore, extensibility of more than 1.5 cm is “positive.” If extensibility >2.0 cm is present especially in combination with other cutaneous features, such as papyraceous scars, molluscoid pseudotumors and/or subcutaneous spheroids, consider other EDS types as possible alternative diagnoses (mainly cEDS and classical-like EDS).
- Piezogenic papules are herniations of subcutaneous fat often demonstrable in the heel upon standing (Fig. 3). It is considered uncommon in children but can be found in adults with history of prolonged standing (occupational), marathon runners, or weightlifters [Poppe and Hamm, 2013] However, in a sex- and age-matched study of 29 Dutch EDS patients, piezogenic papules were found in 34.5% but none in the control group [Kahana et al., 1987].
- Atrophic scarring is defined as scars from linear traumatic lacerations or single-surgery that are unusually shallow (i.e., thin and sunken) and/or wider than the original wound due to impaired repair and subsequent dermal hypotrophy. Atrophic scars as the result of multiple incisions, wound infections, or inflammatory conditions (such as viral infections, cystic acne, etc.) are not to be considered. Elliptical incisions (e.g., for removal of nevi) may be difficult to assess without knowing the size of the original wound. True skin fragility, such as the propensity to have an open wound due to trivial trauma, is not a typical feature of hEDS. Atrophic scarring in hEDS is mildly to moderately different from that usually considered typical of cEDS (Fig. 1).
- Includes history of dental crowding or orthodontic intervention(s) to correct such problems. Both conditions must be positive to meet this criterion.
- Some studies show no increase in the frequency of clinically significant MVP [Dolan et al., 1997; McDonnell et al., 2006; Atzinger et al., 2011] and others show an MVP frequency of 28–67% among hEDS patients [Camerota et al., 2014; Kozanoglu et al., 2016]. This feature is included in the diagnostic criteria because it can be a marker of connective tissue laxity, but is usually not clinically significant in patients with hEDS.
- “Dislocation” is defined as displacement of a bone out of the joint socket (or out of normal position in the case of sesamoid bones such as the patella), sufficiently severe to limit motion of the joint and requiring manual reduction.
- Refers to sites regardless of laterality. For example, right and left patellar instability would count as two. Instability should be evaluated and determined by a qualified practitioner using recommended guidelines.
- All reported aEDS patients had congenital bilateral hip dislocation. One unreported molecularly proven aEDS patient is known to have had congenital unilateral hip dislocation [Byers et al., personal communication].
- For definition of GJH, see criteria for “Classical EDS.”
- Craniofacial features include: prominent and protuberant eyes with puffy, oedematous eyelids and excessive periorbital skin, epicanthal folds, downslanting palpebral fissures, blue sclerae, large fontanels and/or wide cranial sutures, delayed closure of fontanels and hypoplastic chin.
- Reported perinatal complications due to connective tissue fragility include: congenital skull fractures, intracerebral hemorrhage, friable umbilical cord, congenital skin tears, neonatal pneumothorax.
- For definition of GJH, see criteria for “Classical EDS.”
- Most patients identified to date display a severe phenotype, recognizable from birth or first months of life. Milder forms of the condition have recently been described.
- Muscular hypotonia can be very pronounced and lead to delayed gross motor development. This condition should be considered in the initial differential diagnosis of a floppy infant. Neuromuscular work-up is however normal.
- Kyphoscoliosis is usually present at birth or develops in infancy. In patients with biallelic PLOD1mutations, it may be absent throughout adulthood.
- For definitions of GJH and skin hyperextensibility, see criteria for “Classical EDS.”
- Scleral and ocular fragility were removed from the major clinical criteria of kEDS-PLOD1, as rupture of the eye globe following minimal trauma has only been reported in five individuals, including one patient with both eyes affected.
- Facial dysmorphic features include: low-set ears, epicanthal folds, down-slanting palpebral fissures, synophrys, and high palate.
- For definitions of GJH and skin hyperextensibility, see criteria for “Classical EDS.”
- 33 Characteristic craniofacial features associated with biallelic B4GALT7 mutations include: triangular face, wide-spaced eyes, proptosis, narrow mouth, low-set ears, sparse scalp hair, abnormal dentition, flat face, wide forehead, blue sclerae, and cleft palate/bidif uvula.
- Reported radiographic findings associated with biallelic B4GALT7 mutations include: include radioulnar synostosis, metaphyseal flaring, osteopenia, radial head subluxation or dislocation, and short clavicles with broad medial ends.
- Characteristic craniofacial features associated with biallelic B3GALT6 mutations include: midfacial hypoplasia, frontal bossing, proptosis, or prominent eyes, blue sclerae, downslanting palpebral fissures, depressed nasal bridge, long upperlip, low-set ears, micrognathia, abnormal dentition, cleft palate, sparse hair.
- Reported radiographic features associated with biallelic B3GALT6 mutations include: platyspondyly, anterior beak of vertebral body, short ilium, prominent lesser trochanter, acetabular dysplasia, metaphyseal flaring, metaphyseal dysplasia of femoral head, elbow malalignment, radial head dislocation, overtubulation, bowing of long bones, generalized osteoporosis, healed fractures. Craniosynostosis and radioulnar dysostosis has been reported in one patient.
- Reported radiologic findings associated with biallelic SLC39A13 mutations include: mild to moderate platyspondyly, mild to moderate osteopenia of the spine, small ileum, flat proximal femoral epiphyses, short, wide femoral necks.
- Characteristic craniofacial features include: large fontanelle, hypertelorism, short and downslanting palpebral fissures, blue sclerae, short nose with hypoplastic columella, low-set and rotated ears, high palate, long philtrum, thin upper lip vermilion, small mouth, micro-retrognathia.
- For definition of skin hyperextensibility, see criteria for “Classical EDS.”
- The phenotypic features in the three reported patients with EDS caused by DSE deficiency seem to be milder than those in patients with EDS caused by D4ST1-deficiency, but identification of additional patients with DSE-deficiency is needed to confirm this correlation.
- So far, five families have been reported: four with an autosomal dominant condition and one with an autosomal recessive condition. The affected siblings from the family with the autosomal recessive condition have a more severe form of the condition than patients with autosomal dominant inheritance [Zou et al., 2014].
- Muscle biopsy and skin fibroblast culture studies: the diagnosis can be suspected in patients that undergo a muscle biopsy and/or in whom a fibroblast line is established. In the autosomal recessive form in which there is no collagen XII produced, immunostaining has shown absence of collagen XII staining. In missense mutations that lead to autosomal dominant forms, collagen XII may be abnormally secreted. The myopathic pattern on muscle biopsy may be suggestive, but is not diagnostic. Recently, muscle MRI has been developed as an alternative, non-invasive technique to study muscle involvement, however it is not specific enough to confirm the diagnosis.
Quelle:
Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):8-26. doi: 10.1002/ajmg.c.31552. PMID: 28306229.
