Krankheitsbild
Ehlers-Danlos-Syndrome und
Hypermobilitäts-Spektrum-Erkrankung

Hypermobiles Ehlers-Danlos-Syndrom [hEDS] und Hypermobilitäts-Spektrum-Erkrankung [Hypermobility Spectrum Disorders, HSD]
Kenneth S. Yew, MD, MPH; Kara A. Kamps-Schmitt, MD; und Robyn Borge, MD
Gundersen Medical Foundation Family Medicine Residency, La Crosse, Wisconsin
Das hypermobile Ehlers-Danlos-Syndrom [hEDS] und die Hypermobilitäts-Spektrum-Erkrankung [Hypermobility Spectrum Disorders, HSD] sind die am meist häufigsten symptomatischen Gelenkhypermobilitätszustände, die in der klinischen Praxis vorkommen. Die internationale Klassifikation der Ehlers-Danlos-Syndrome von 2017 ersetzt frühere Bezeichnungen für symptomatische Gelenkhypermobilität durch hEDS und führt den Begriff HSD für Patientinnen und Patienten ein, die die Diagnosekriterien für hEDS nicht erfüllen. Beide werden anhand der Diagnosekriterien von 2017 diagnostiziert, die auch andere, weniger häufige Erkrankungen mit Gelenkhypermobilität ausschließen, wie z.B. andere Formen der Ehlers-Danlos-Syndrome und andere vererbbare Bindegewebserkrankungen. hEDS kann einem autosomal-dominanten Vererbungsmuster folgen, aber es gibt keine bekannte ursächliche Genveränderung, die bei der Diagnose helfen könnte. Zu den klinischen Merkmalen des hEDS gehören die Gelenkhypermobilität, Hautbefunde und Gelenkschmerzen oder rezidivierende Luxationen. hEDS sowie seltener auch HSD, können mit verschiedenen extraartikulären Symptomen einhergehen, es können Angststörungen, chronische Schmerzen, Fatigue, orthostatische Intoleranz, funktionelle gastrointestinale Störungen sowie Becken- und Blasenfunktionsstörungen auftreten. Die zentralen Ziele der Therapie sind die Behandlung der Symptome, die Vorbeugung von Gelenkschäden sowie die Aufklärung der Patientinnen und Patienten über ihre Erkrankung. Auf der Grundlage begrenzter Erkenntnisse können Betroffene mit hEDS/HSD von Physio- und Ergotherapie, psychologischer Unterstützung und Selbstmanagement profitieren. Eine Schlüsselrolle spielen Hausärztinnen und Hausärzte nicht nur bei dem ersten Verdacht einer Diagnose, sondern auch bei der Koordination eines multidisziplinären Teams, welches viele Betroffene für ihre Behandlung benötigen. (Am Fam Physician. 2021;103(8):481-492. Copyright © 2021 American Academy of Family Physicians.)
Hypermobiles Ehlers-Danlos-Syndrom [hEDS] und die Hypermobilitäts-Spektrum-Erkrankung [Hypermobility Spectrum Disorders, HSD] sind die häufigsten symptomatischen Gelenkhypermobilitätszustände, die in der klinischen Praxis auftreten.1,2 Hausärztinnen und Hausärzte spielen eine wichtige Rolle bei der Versorgung von Betroffenen mit diesen Erkrankungen, von der Erstdiagnose bis zur weiterführenden Betreuung.
Definitionen
„Ehlers-Danlos-Syndrome (EDS) ... sind eine Gruppe von erblichen Bindegewebserkrankungen, die durch Anomalien in der Struktur, Produktion und/oder Verarbeitung von Kollagenen verursacht werden. Die neue Klassifikation aus dem Jahr 2017 umfasst 13 Untertypen von EDS.“3 Tabelle 1 beschreibt diese Untertypen.1,4,5 Die Gelenkhypermobilität ist ein gemeinsames Merkmal vieler EDS-Subtypen und anderer erblicher Bindegewebserkrankungen. Die Gelenkhypermobilität ist definiert als die Fähigkeit, ein Gelenk „über den normalen Bewegungsradius hinaus -aktiv oder passiv- zu bewegen“.4 Die Gelenkhypermobilität kann einige wenige oder viele Gelenke betreffen und kann völlig symptomlos verlaufen.
Zusätzliche Inhalte unter https://www.aafp.org/afp/2021/0415/p481.html.
CME Dieser klinische Inhalt entspricht den AAFP-Kriterien für CME. Siehe CME-Quiz auf Seite 460.
Offenlegung der Autorenschaft: Keine relevanten finanziellen Zuwendungen.
Patienteninformation: Ein Handout zu diesem Thema, das von der Autorenschaft dieses Artikels verfasst wurde, ist unter https://www.aafp.org/afp/2021/0415/p481-s1.html verfügbar.
NEUES ZU DIESEM THEMA
Hypermobilitäts-Spektrum-Erkrankung
In der internationalen Klassifikation der Ehlers-Danlos-Syndrome von 2017 wurden frühere Bezeichnungen für symptomatische Gelenkhypermobilität durch hypermobiles Ehlers-Danlos-Syndrom ersetzt und der Begriff Hypermobilitäts-Spektrum-Erkrankung für Betroffene eingeführt, die die Diagnosekriterien des hypermobilen Ehlers-Danlos-Syndroms nicht erfüllen.
Eine Bevölkerungsumfrage in Großbritannien aus dem Jahr 2013 ergab, dass 3,4 % der Erwachsenen unter einer Hypermobilität sowie chronischen Schmerzen am Bewegungsapparat leiden.
Die generalisierte Gelenkhypermobilität tritt häufiger auf als das hypermobile Ehlers-Danlos-Syndrom/ Hypermobilits-Spektrum-Erkrankung. Aber Betroffene mit generalisierter Gelenkhypermobilität können asymptomatisch sein. Bei der Untersuchung von Schülerpopulationen anhand der Kriterien von 2017 wiesen 4 % bis 11 % der Kinder und Jugendlichen im Alter von 3 bis 19 Jahren eine generalisierte Gelenkhypermobilität auf.
Tabelle 1
Überschneidungen zwischen Gelenkhypermobilität, Hypermobilitäts-Spektrum-Erkrankung und Ehlers-Danlos-Syndromen
|
Typ |
Beighton-Score |
Muskuloskelettale Beteiligung* |
Anmerkungen |
|
Asymptomatische Gelenkhypermobilität |
|||
|
Asymptomatische generalisierte Gelenkhypermobilität |
Positiv |
Fehlend |
- |
|
Asymptomatische periphere Gelenkhypermobilität |
Normalerweise negativ |
Fehlend |
Die Gelenkhypermobilität beschränkt sich typischerweise auf Hände und/oder Füße |
|
Asymptomatische lokalisierte Gelenkhypermobilität |
Negativ |
Fehlend |
Die Gelenkhypermobilität beschränkt sich auf einzelne Gelenke oder Körperteile |
|
Hypermobilitäts-Spektrum-Erkrankung [HSD] |
|||
|
Generalisierte HSD |
Positiv |
Vorhanden |
Erfüllt NICHT die Kriterien für ein hEDS, basierend auf begrenzten Befunden in der Haut und im muskuloskelettalen System sowie einer fehlenden Familienanamnese Keine Gene identifiziert Echokardiographie-Screening ist nicht erforderlich |
|
Peripheres HSD |
Normalerweise negativ |
Vorhanden |
Die Gelenkhypermobilität ist typischerweise auf Hände und/oder Füße beschränkt |
|
Lokalisiertes HSD |
Negativ |
Vorhanden |
Die Gelenkhypermobilität beschränkt sich auf einzelne Gelenke oder Körperteile |
|
Historisches HSD |
Negativ |
Vorhanden |
Die Gelenkhypermobilität war in der Vergangenheit nachweisbar |
|
EDS - Gelenkhypermobilität mit ausgeprägteren Haut- und Muskelskelettbefunden und/oder positiver Familienanamnese |
|||
|
1. Hypermobiles EDS [hEDS] |
Positiv |
Möglich |
Die Kriterien sind aufgrund von unterstützenden Befunden bei Haut und Körpersystemen und/oder positiver Familienanamnese erfüllt (siehe Abbildung 2) Keine Gene identifiziert AD - Vererbungsmuster Echokardiographie-Screening ist erforderlich |
|
Typ |
Beighton-Score |
Majorkriterien |
Betroffene Gene |
|
2. Klassisches EDS [cEDS] |
Positiv |
Überdehnbarkeit der Haut Abnorme Narbenbildung |
COL5A1-, COL5A2-Gene selten COL1A1-Gen AD-Vererbung |
|
3. Classical-like EDS [clEDS] |
Positiv |
Überdehnbarkeit der Haut Neigung zu Hämatomen |
TNXB-Gene AR - Vererbung |
|
4. Kardio-valvuläres EDS [cvEDS] |
Positiv oder negativ, generalisierte Hypermobilität oder beschränkt auf kleine Gelenke |
Herzklappenanomalien Hautveränderungen |
COL1A2-Gen AR-Vererbung |
|
5. Vaskuläres EDS [vEDS] |
Positiv oder negativ |
Familienanamnese von vaskulärem EDS Frühzeitige arterielle Ruptur oder Uterusruptur, Perforation des Colon sigmoideum oder atraumatische Fistelbildung im Sinus carotis-cavernosus |
COL3A1-Gen seltene COL1A1-Gen AD-Vererbung |
|
6. Arthrochalasie EDS [aEDS] |
Positiv |
Angeborene bilaterale Hüftluxationen Hyperextensibilität der Haut |
COL1A1, COL1A2 Gene AD-Vererbung |
|
7. Dermatosparaxis EDS [dEDS] |
Positiv oder negativ |
Extreme Fragilität der Haut Charakteristische kraniofaziale Merkmale |
ADAMTS2 Gen AR-Vererbung |
|
8. Kyphoskoliotisches EDS [kEDS][ |
Positiv mit Luxationen und Subluxation in der Vorgeschichte |
Kongenitale Hypotonie Kyphoskoliose |
PLOD1, FKBP14 Gene AR-Vererbung |
|
9. Brittle cornea syndrome [BCS] |
Positiv oder negativ |
Dünne Hornhaut mit oder ohne Ruptur Keratokonus Keratoglobus Blaue Skleren |
ZNF469, PRDM5 Gene AR-Vererbung |
|
10. Spondylodysplastisches EDS [spEDS] |
Positiv oder negativ |
Kleinwuchs Muskelhypotonie Beinachsenfehlstellungen |
B4GALT7, B3GALT6, SLC39A13 Gene AR-Vererbung |
|
11. Musculocontraktuelles EDS [mcEDS] |
Positiv oder negativ |
Angeborene multiple Kontrakturen Charakteristische kraniofaziale Merkmale Beteiligung der Haut |
CHST14, DSE Gene AR-Vererbung |
|
12. Myopathisches EDS [mEDS] |
Distale Gelenke betroffen |
Angeborene Muskelhypotonie und/oder -atrophie, die sich mit zunehmendem Alter verbessern Proximale Muskelkontrakturen |
COL12A1 Gen AD- oder AR-Vererbung |
|
13. Periodontales EDS [pEDS] |
Positiv oder negativ |
Parodontitis Fehlende befestigte Gingiva Prätibiale Plaques Periodontales EDS in der Familiengeschichte |
C1R, C1S Gene AD-Vererbung |
AD = autosomal dominant; AR = autosomal rezessiv; EDS = Ehlers-Danlos-Syndrome.
Informationen aus den Referenzen 1, 4 und 5 und persönliche Kommunikation von Karyn Laursen, MD.
*-Die muskuloskelettale Beteiligung umfasst Folgendes: (1) Schmerzen; (2) Muskuloskelettale-/Weichteiltraumata, einschließlich Luxationen, Subluxationen, Weichteilschäden und Mikrotraumata (Mikrotraumata umfassen kleine Muskelrisse, verstauchte Bänder, gezerrte Muskeln und überdehnte Sehnen); (3) Störung der Propriozeption; und (4) andere Erkrankungen des Bewegungsapparates (z.B. Plattfüße; Valgus Anomalie des Ellenbogens, des Rückfußes und des Hallux; Kyphose; Skoliose; Plagiozephalie).
Im Unterschied zu einer lokalisierten Gelenkhypermobilität ist eine generalisierte Gelenkhypermobilität eher mit einem genetischen Syndrom verbunden. Das hEDS stellt die häufigste EDS-Variante dar und macht 80 % bis 90 % der EDS-Fälle aus. Zu den klinischen Merkmalen gehören Gelenkhypermobilität, Hautbefunde (Abbildung 1) und Gelenkschmerzen oder rezidivierende Luxationen (Abbildung 26 und Abbildung 31,7). Mit der Klassifikation von 2017 wurden strengere Kriterien für hEDS eingeführt, um Betroffene mit einem hEDS von denjenigen zu unterscheiden, bei denen am ehesten ein genetisch diagnostizierbares Syndrom vorliegt.1 Für Betroffene mit symptomatischer Gelenkhypermobilität, die weder die neuen hEDS-Kriterien noch eine andere spezifische Erkrankung erfüllen, werden mit der Klassifikation von 2017 die HSD eingeführt.1
Abbildung 1

Atrophische Narbe
Abbildung 2
Die Urheberrechte aus dem Originalartikel für die Abbildung 2 liegen uns nicht vor.
[In Zusammenarbeit mit Herrn Dr. Pradeep Chopra wurde eine ausfüllbare Checkliste zur Diagnostik des hypermobilen Ehlers-Danlos-Syndromes (hEDS) erstellt, die über folgenden Link heruntergeladen werden kann: Klinische Checkliste für die Diagnostik des hypermobilen Ehlers-Danlos-Syndromes (hEDS)]
Betroffene mit einem HSD unterscheiden sich von denen mit einem hEDS und anderen Syndromen mit Gelenkhypermobilität dadurch, dass ihre Symptome primär muskuloskelettal sind. Eine extraartikuläre Beteiligung kann aber auch begrenzt auftreten.4
Alle früheren Bezeichnungen, einschließlich EDS Typ III, EDS-Hypermobilitätstyp, Hypermobilitätssyndrom, Gelenkhypermobilitätssyndrom und gutartiges Gelenkhypermobilitätssyndrom, sollten nicht mehr verwendet werden.4 Man ging früher davon aus, dass es sich bei diesen genannten Diagnosen, um unterschiedliche Entitäten handelte. Doch in späteren Studien zeigte sich, dass eine breite Überschneidung dieser älteren benannten Erkrankungen in den betroffenen Familien feststellbar waren. Dabei zeigte sich, dass es sich um die ein und dieselbe Entität handelte.4 In einer neueren Studie erfüllten fast alle Betroffene mit einer der früheren, aber inzwischen überholten Diagnosen, entweder die Kriterien für ein hEDS oder für die HSD.8 Da es sich hierbei um die häufigsten symptomatischen Hypermobilitätszustände handelt, stehen ihre Bewertung und Behandlung im Mittelpunkt dieses Artikels. Die Begriffe hEDS und HSD werden verwendet, es sei denn, es erfordert einen Verweis auf einen älteren Diagnosebegriff.
Epidemiologie und Pathogenese
Die genaue Prävalenz von hEDS/HSD ist nicht bekannt. Die besten Einschätzungen der Prävalenz dieser Erkrankungen in der Bevölkerung stammen aus Studien von nationalen Registern oder Patientenregistern aus Schweden, Wales und Großbritannien. Dabei wurden Diagnoseschlüssel für EDS und den Gelenkhypermobilitätssyndromen verwendet, wobei letzteres ein älterer Begriff für hEDS ist.9,10 Die in diesen beiden Studien für alle EDS und Gelenkhypermobilitätssyndrome zusammen kodiert festgestellte kombinierte Prävalenz von hEDS/HSD war niedriger als 0,13 % bis 0,19 % (7-10 Betroffene/5000 Personen).9,10 Eine weitere Einschätzung der kombinierten Prävalenz von hEDS/HSD aus einer britischen validierten Bevölkerungsumfrage ergab, dass 3,4 % der Erwachsenen eine Hypermobilität sowie chronische Schmerzen im Bewegungsapparat angaben.11
Eine generalisierte Gelenkhypermobilität ist ein diagnostisches Kriterium für hEDS. Dies kommt weitaus häufiger vor als hEDS/HSD, da Betroffene mit generalisierter Gelenkhypermobilität asymptomatisch sein können. Bei der Untersuchung von Schülerpopulationen anhand der Kriterien von 2017, wiesen 4 bis 11 % der Kinder und Jugendlichen im Alter von 3 bis 19 Jahren eine generalisierte Gelenkhypermobilität auf.12-17 Der Prozentsatz der diagnostizierten Betroffenen mit generalisierter Gelenkhypermobilität [hEDS/HSD] ist unbekannt.
hEDS ist der einzige EDS-Subtyp, für den noch keine genetische Mutation entdeckt wurde. Man geht davon aus, dass hEDS autosomal-dominant mit unvollständiger Penetranz vererbt wird. Die Pathogenese des hEDS/HSD ist noch nicht vollständig entschlüsselt. Diese beinhalten jedoch eine Laxität der Muskeln und Sehnen,18 eine verminderte Propriozeption,19 eine erheblich gestörte Bindegewebsstruktur sowie Veränderungen der Genexpression.20
Abbildung 3
Gelenkhypermobilität deutet auf aktuelle oder frühere Gelenkinstabilität, (Sub)Luxationen oder Überbeweglichkeit hin
und
| • Hautbefunde wie weiche/samtige Haut, atrophische Narben, Überdehnbarkeit der Haut, unerklärliche Striae oder • Chronische muskuloskelettale Schmerzen oder |
• Rezidivierende Hernien, Beckenorgan- oder Rektumprolpas oder • Marfanoider Habitus oder • Familienanamnese von EDS |
|
|
|
|
|
|
|
|
|
|
|
|
|
▼ |
|
|
|
|
|
Wenn der Beighton-Score einen Punkt unter dem altersspezifischen |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
▼ |
|
|
|
|
|
Hat die Patientin oder der Patient eine generalisierte Gelenkhypermobilität? |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
Nein |
|
|
|
Ja |
|
|
|
▼ |
|
|
▼ |
|
||
|
Hat die Patientin oder der Patient ein Hypermobilitäts Spektrum Erkrankung [HSD]? |
Anwendung der übrigen Diagnosekriterien für hypermobiles EDS [hEDS], einschließlich der Durchführung einer Echokardiographie |
||||||
|
|
|
|
|
|
|
|
|
|
|
Nein |
|
Ja |
|
|
|
|
|
|
|
▼ |
|
▼ |
|
||
|
|
Ärtzliche Kontrollen mit therapeutischen Behandlungen, sowie geeigneten pflegerischen Unterstützungen, um auf eine evtl. Entwicklung eines hypermobilen EDS oder einer anderen vererbbaren Bindegewebsstörung hinzuweisen |
Merkmale, die auf andere vererbbare oder erworbene Bindegewebsstörungen hinweisen? |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nein |
|
Ja |
|
|
|
|
|
▼ |
▼ |
||
|
|
|
|
|
Hat die Patientin oder der Patient ein hypermobiles EDS? |
Nach Bedarf weitere Untersuchungen durchführen (z. B. weitere bildgebende Verfahren, Labortests, Augenuntersuchungen, genetische Untersuchungen) |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nein |
|
Ja |
|
|
▼ |
|
▼ |
▼ |
|
|||
|
Patientin oder den Patienten nach klinischer Indikation behandeln und die Veränderungen der Symptomatik beobachten |
Behandlung nach klinischer Indikation |
||||||
Kriterien für generalisierte Gelenkhypermobilität*
- Beighton-Score ≥ 6 bei präpubertären Kindern und Jugendlichen
- Beighton-Score ≥ 5 ab der Pubertät bis zum Alter von 50 Jahren
- Beighton-Score ≥ 4 bei Personen, die älter als 50 Jahre sind
Wenn der Fünf-Punkte-Fragebogen positiv ist (d.h. zwei oder mehr Ja-Antworten), kann ein Punkt hinzugefügt werden
Fünf-Punkte-Fragebogen†
Der Fünf-Punkte-Fragebogen ist positiv, wenn die Patientin oder der Patient zwei oder mehr Fragen mit Ja beantwortet
Können Sie jetzt (oder konnten Sie jemals) Ihre Hände flach auf den Boden legen, ohne die Knie zu beugen?
Können Sie jetzt (oder konnten Sie jemals) Ihren Daumen beugen, um Ihren Unterarm zu berühren?
Konnten Sie als Kind Ihre Freunde belustigen, indem Sie Ihren Körper in seltsame Positionen gebracht haben, oder konnten Sie einen Spagat machen?
Haben Sie sich als Kind oder Jugendlicher mehr als einmal die Schulter oder die Kniescheibe (sub)luxiert?
Halten Sie sich für überbeweglich? Diagnostisches Vorgehen bei einer Patientin oder einem Patienten mit einem möglichen hypermobilen EDS.
EDS = Ehlers-Danlos-Syndrome
Diagnostisches Vorgehen bei einer Patientin oder einem Patienten mit einem möglichen hypermobilen EDS.
Informationen aus den Referenzen 1 und 7.
Klinische Präsentation
hEDS und HSD weisen ein komplexes Spektrum von Anzeichen und Symptomen unterschiedlichen Ausmaßes und unterschiedlicher Kombinationen auf. Diese machen es schwierig, diese Erkrankungen zu erkennen. Die allgemeinen Merkmale des hEDS sind in Tabelle 2 aufgeführt.1,2,21 Die Prävalenz der generalisierten Gelenkhypermobilität nimmt mit dem Alter ab2. Dieser Rückgang wird in den Kriterien für hEDS von 2017 berücksichtigt, indem die Anamnese für Patientinnen und Patienten mit einer unterschwelligen Gelenkhypermobilität aufgenommen wurden.1,7 Zu den ausgeprägtesten systemischen Befunden, die mit einem hEDS assoziiert sind, können Angststörungen, chronische Schmerzen, Fatigue, orthostatische Intoleranz, funktionelle gastrointestinale Störungen sowie Becken- und Blasenfunktionsstörungen auftreten.22-26 Tabelle 3 beschreibt die Symptome und die körperlichen Befunde, die häufig mit einem hEDS in Verbindung gebracht werden.2,22,23,26-38
Diagnostische Evaluierung
KLINISCHE MERKMALE
Die Diagnose eines hEDS sollte bei Patientinnen und Patienten mit den in Tabelle 2 aufgeführten klinischen Merkmalen in Betracht gezogen werden.1,2,21 Bei Patientinnen und Patienten mit systemischen Manifestationen (Tabelle 32.22,23,26-38) und einer Vorgeschichte von Gelenkhypermobilität oder Arthralgien, kann ebenfalls ein hEDS oder eine verwandte Erkrankung vorliegen, die übersehen oder zuvor falsch diagnostiziert wurde.1 Eine Befragung unter britischen Betroffenen ergab, dass die durchschnittliche Zeit bis zur Diagnose 10 Jahre beträgt.39
Die Diagnose des hEDS/HSD wird anhand der Anamnese, der körperlichen Untersuchung und des Ausschlusses anderer Erkrankungen gestellt, die mit einer muskuloskelettalen Hypermobilität einhergehen.2,40,41 Die Diagnosekriterien für ein hEDS sind in Abbildung 2 aufgeführt.6 Tabelle 1 enthält die Kriterien für eine HSD, wenn Patientinnen und Patienten weder die Kriterien für ein hEDS noch eine andere spezifische Erkrankung erfüllen.1,4,5 Eine Differenzialdiagnose für Gelenkhypermobilität findet sich in Tabelle 4.1,4,21,42
Tabelle 2
Häufige Erscheinungsformen des hypermobilen Ehlers-Danlos-Syndroms
|
Merkmale |
|
Anmerkungen |
|
Vorgeschichte* |
|
|
|
Extreme Flexibilität oder Überbeweglichkeit |
Ältere Erwachsene können sich vielleicht daran erinnern, dass sie in ihrer Kindheit überbeweglich oder extrem flexibel waren |
|
|
Rezidivierende oder chronische Gelenkschmerzen |
Können zeitweise oder chronisch auftreten |
|
|
Gelenksubluxation oder -luxationen ohne signifikantes Traumata |
Schulter, Knie und Hüfte sind am häufigsten betroffen |
|
|
Rezidivierende Hernien, Beckenorganprolaps oder Rektumprolaps |
Vor allem, wenn keine andere bekannte prädisponierende Erkrankung vorliegt |
|
|
Körperlicher Befund |
|
|
|
Hautbefund |
|
*Merkmale können nacheinander auftreten.
Informationen aus den Referenzen 1, 2 und 21.
Tabelle 3
Symptome im Zusammenhang mit dem hypermobilen Ehlers-Danlos-Syndrom
|
Organsysteme |
Symptome/körperliche Befunde |
|
Autonomes Nervensystem27,28 |
Neural vermittelte Hypotonie/Synkope |
|
Kardiovaskulär29 |
Gering progrediente Aortenwurzeldilatation |
|
Gastrointestinal22 |
Chronische/rezidivierende Gastritis |
|
Gynäkologisch26,30 |
Zyklusbedingte Dysmenorrhöe |
|
Mukokutan31 |
Atrophische Narben |
|
Organsysteme |
Symptome/körperliche Befunde |
|
Muskuloskelettal2,32-34 |
Chronische Schmerzen |
|
Neurologisch32,34,35 |
Ungeschicklichkeit |
|
Auge36 |
Myopie und/oder Strabismus |
|
Psychologisch23,37,38 |
Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung [ADHS] |
[*Redaktionelle Anmerkung: Bei ME/CFS handelt es sich NICHT um eine psychische Erkrankung. Die Weltgesundheitsorganisation (WHO) klassifiziert ME/CFS als neurologische Erkrankung (ICD-10 G93.3). Quelle: https://www.aerzteblatt.de/archiv/231286/Myalgische-Enzephalomyelitis-Chronisches-Fatigue-Syndrom-Interdisziplinaer-versorgen]
Informationen aus den Referenzen 2, 22, 23 und 26-38.
Eine sorgfältige Familienanamnese, in der nach Gelenkhypermobilität, muskuloskelettalen Symptomen, Aneurysmen und genetischen Erkrankungen gefragt wird, ist unerlässlich. Sobald der Verdacht auf ein hEDS besteht, sollte die Ärztin oder der Arzt den Grad und das Ausmaß der Hypermobilität mithilfe eines Beighton-Score1,40,41,43 (Tabelle 51,44) bestimmen. Der Beighton-Score umfasst fünf diagnostische Untersuchungen, um einen Score zwischen 0 und 9 zu berechnen. Die Kriterien für ein hEDS von 2017 in Abbildung 2 legen fest, falls der Beighton-Score einen Punkt unter den alters- und geschlechtsspezifischen Grenzwerten für eine generalisierte Gelenkhypermobilität liegt, dass als nächster Schritt der validierte fünfteilige Fragebogen verwenden werden sollte. Es muss festgestellt werden, ob die Patientin oder der Patient eine generalisierte Gelenkhypermobilität aufweist. Dies ist ein notwendiges Kriterium für die Diagnose des hEDS.6,7 Patientinnen und Patienten ohne generalisierte Gelenkhypermobilität können dennoch an einem HSD leiden. In Abbildung 3 wird eine Bewertungsstrategie für Patientinnen und Patienten vorgeschlagen, bei denen der Verdacht auf ein hEDS oder ein HSD besteht.1,7
Wenn sich die generalisierte Gelenkhypermobilität bei Patientinnen und Patienten mit Verdacht auf ein hEDS bestätigt, werden die übrigen Kriterien für hEDS abgefragt1 (Abbildung 26). Dazu gehört die Anamnese der Patientinnen und Patienten nach muskuloskelettalen Symptomen, abdominalen Hernien sowie Organ- und Mitralklappenprolaps, die Untersuchung der Haut, die Prüfung auf Arachnodaktylie und die Messung des Verhältnisses von Armspannweite zu Körpergröße.
Abbildung 1 zeigt eine typische atrophische Narbe.
DIAGNOSETEST
Da es keinen Bestätigungstest gibt, bleiben das hEDS und die HSD klinische Diagnosen.2 Labortests und radiologische Befunde zur Feststellung einer erworbenen Bindegewebsstörung oder eines Verdachts auf eine Knochen- oder Gelenkverletzung richten sich nach der Anamnese. Das Vorhandensein von marfanoiden Merkmalen erfordert eine Unterscheidung zwischen hEDS und Marfan-verwandten Syndromen. In Tabelle 4 sind Merkmale aufgeführt, die bei der Unterscheidung zwischen diesen Erkrankungen weiterhelfen können.1,4,21,42 Bei Patientinnen und Patienten mit einem möglichem hEDS sollte ein Echokardiographie-Screening durchgeführt werden, um eine Aortenwurzeldilatation oder einen Mitralklappenprolaps festzustellen. Spezifische genetische Untersuchungen sollten bei einem Verdacht auf andere EDS-Varianten, dem Marfan Syndrom, dem Loeys-Dietz-Syndrom sowie andere genetische Erkrankungen durchgeführt werden (Tabelle 4).1,4,21,42 Oftmals sind mehrere ärztliche Termine erforderlich, um eine diagnostische Bewertung durchzuführen. Viele Patientinnen und Patienten, die die Kriterien für ein hEDS nicht erfüllen und keine eindeutigen Hinweise auf ein anderes spezifisches Syndrom aufweisen, erfüllen die Kriterien für HSD. Bleibt die Diagnose unklar, kann eine Überweisung an ein genetisches Zentrum zur weiteren Abklärung erforderlich sein. eTabelle A enthält eine Liste zur Unterstützung der Diagnose und Behandlung von Hypermobilitätssyndromen.
Management
Die zentralen Ziele der Therapie sind die Behandlung der Symptome, die Vorbeugung von Gelenkverletzungen und die Aufklärung der Betroffenen über ihre Erkrankung.45-48 Basierend auf begrenzter Evidenz und Expertenmeinungen umfassen die Hauptpfeiler der Behandlung von hEDS und HSD die Aufklärung der Betroffenen, Physio- und Ergotherapie, psychologische Unterstützung und Selbstmanagement. Die Symptome von HSD können mit der Therapie abklingen, fortbestehen oder sich zu einem hEDS entwickeln. hEDS wird als lebenslanger Zustand angesehen, da es derzeit keine heilende Behandlung gibt.
Aufgrund der multisystemischen Beteiligung, ergeben sich unterschiedliche Behandlungsstrategien. Betroffene mit einem hEDS können von einem multidisziplinären Team profitieren, zu dem Ärztinnen und Ärzte, Pflegepersonal, Physio- und Ergotherapierende, Orthopädietechnikerinnen und Orthopädietechniker, Ernährungsberatende, Lifestyle-Coaches, psychologische aber auch Community- und Online-Unterstützung gezählt werden können.
Tabelle 4
Ausgewählte Differenzialdiagnosen der Gelenkhypermobilität
|
Befunde |
|
Unterscheidungsmerkmale |
|
Erworbene Erkrankungen wie diffuse degenerative Störungen der Muskeln, Gelenke oder Nerven, Hypothyreose oder Unterernährung |
Aussagekräftige Anamnese |
|
|
Chromosomale und genomische Störungen einschließlich Down-Syndrom, Aneuploidien (47, XXY; 47, XXX) und verschiedene Mikrodeletionen- und Mikroduplikationssyndrome |
Dysmorphien-Hypogonadismus |
|
|
Hereditäre Cutis Laxa42: mehrere Subtypen |
Schlaffe, unelastische Haut |
|
|
Hereditäre Myopathien: Bethlem-, Ullrich und andere Myopathien |
Hypotonie und Fatigue |
|
|
Loeys-Dietz-Syndrom |
Arterielle Tortuosität und Aortenaneurysmen, Lippen-Kiefer-Gaumenspalte |
|
|
Marfan-Syndrom, Beals-Syndrom, MASS-Phänotyp und Arterial-Tortuosity Syndrom |
Ectopia lentis beim Marfan-Syndrom |
|
|
Mehrfache kongenitale Anomalien oder geistige Behinderungen, einschließlich RASopathien, Kabuki-Make-up-Syndrom und FG-Syndrom
|
Mehrfache angeborene Anomalien oder geistige Behinderungen |
|
|
Fragile-X-Syndrom |
||
|
Andere Varianten der Ehlers-Danlos-Syndrome, einschließlich cEDS, vEDS und seltenere Formen |
Akrogerie* |
|
|
Skelettdysplasien: Osteogenesis imperfecta Typ I, Larsen-Syndrom, Desbuquois-Syndrom und andere |
Blaue Skleren |
MASS = Veränderungen an der Mitralklappe und Aorta, Myopie, Haut und Skelettbeteiligung
*-Akrogerie: atrophe Haut und subkutanes Fett an Händen und Füßen, die den Anschein eines beschleunigten Alterns erwecken, wie sie auch beim vaskulären Ehlers-Danlos-Syndrom vorkommen kann.
Informationen aus den Referenzen 1, 4, 21 und 42.
Eine multidisziplinäre Versorgung kann bei der Behandlung von Haut-, Gelenk-, Herz-Kreislauf- und Magen-Darm-Komplikationen sowie chronischen Schmerzen helfen. Hausärztinnen und Hausärzte spielen eine Schlüsselrolle bei der Betreuung und Koordinierung der komplexen Versorgung, die viele Betroffene mit hEDS benötigen.
Die Behandlung von muskuloskelettalen Beschwerden umfasst konservative Behandlung wie: körperliche Aktivität, Paracetamol und nichtsteroidale entzündungshemmende Medikamente [NSAR], Wärme- und/oder Kälteanwendungen, eine verbesserte Ergonomie und Körperhaltung, Entspannungstechniken, Massagen, Hydrotherapie und Gelenkstabilisierungstechniken mit Orthesen und/oder Taping.47,49,50 Medikamente, die die Thrombozytenfunktion beeinträchtigen, sollten bei Betroffenen mit einem hEDS, die eine erhöhte Hämatomneigung aufweisen, generell vermieden werden. Physiotherapierende sollten ihr Augenmerk auf Kräftigungsübungen, propriozeptive Übungen sowie den Schutz der Gelenke legen.51 Ergotherapierende können dazu beitragen, die Muskeln der oberen Extremitäten und der Hände zu stärken, die Aktivitäten des täglichen Lebens zu verbessern und die Betroffenen mit adaptiven Schreibgeräten und anderen weiteren Hilfsmitteln vertraut zu machen. Tai Chi hat sich bei einer Osteoarthritis, Fibromyalgie und bei Kreuzschmerzen als vorteilhaft erwiesen,52 obwohl es bei Betroffenen mit hEDS/HSD noch nicht untersucht wurde. Der Einsatz von Schienen und Orthesen kann bei bestimmten Betroffenen hilfreich sein.
Die Aufklärung der Betroffenen über die Änderungen ihres Lebensstils, Behandlungsmöglichkeiten und Erwartungen, ist eine der wichtigsten Maßnahmen. Die Optimierung des Schlafs, der Schutz der Gelenke durch ein angemessenes Maß an regelmäßiger körperlicher Betätigung (mit geringer Belastung und geringem Widerstand), die Gewichtskontrolle, die Vermeidung von Drogenkonsum (z. B. Alkohol, Nikotin) und eine gesunde Ernährung können Schmerzen, Verletzungen und Müdigkeit verringern, sowie die Mobilität und Funktionalität fördern. Orthostatische Intoleranz kann durch vermehrte Flüssigkeits- und Salzzufuhr und durch das Tragen von Kompressionsstrümpfen gemildert werden.
Tabelle 5
Beighton-Score
|
Diagnostische Untersuchung |
Abbildung |
Bewertung |
Bewertung |
|
Fähigkeit zur passiven Dorsalflexion des kleinen Fingers ≥ 90 Grad
|
|
_ / 1 Punkt |
_ / 1 Punkt |
|
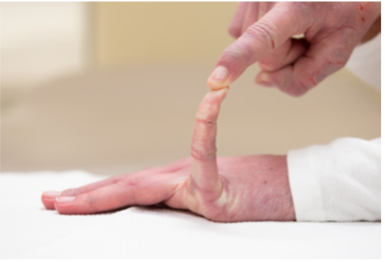
Fähigkeit, den Daumen dem volaren Aspekt des ipsilateralen Unterarms entgegenzusetzen
|
|
_ / 1 Punkt |
_ / 1 Punkt |
|
Fähigkeit zur Überstreckung des Ellenbogengelenks > 10 Grad
|
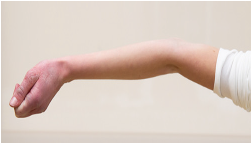 |
_ / 1 Punkt |
_ / 1 Punkt |
|
Fähigkeit zur Hyperextension des Kniegelenks > 10 Grad
|
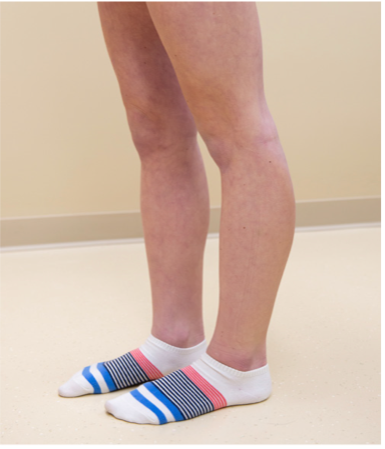 |
_ / 1 Punkt |
_ / 1 Punkt |
|
Fähigkeit, die Händinnenflächen auf den Boden zu legen, indem man sich mit vollständig gestreckten Knien nach vorne beugt
|
 |
_ / 1 Punkt |
|
| Total _ / 9 Punkte | |||

Anmerkung: Der Beighton-Score ist die Gesamtsumme für jede Extremität und die Vorwärtsbeugung.
Informationen aus den Referenzen 1 und 44.
SORT*: WICHTIGE EMPFEHLUNGEN FÜR DIE PRAXIS
|
Klinische Empfehlungen |
Evidenz-bewertung |
Kommentare |
|
Verdacht auf ein hEDS/HSD bei Patientinnen und Patienten mit Gelenkhypermobilität und damit verbundenen Symptomen wie Gelenkschmerzen oder (Sub-)Luxationen und typischen Hautbefunden, Arthralgien, wiederkehrenden Hernien, marfanoidem Habitus oder EDS in der Familienanamnese.1 |
C |
Expertenmeinungen aus der Internationalen Klassifikation der Ehlers-Danlos-Syndrome 2017 |
|
Beurteilung der Gelenkhypermobilität bei Patientinnen und Patienten mit Verdacht auf ein hEDS/ HSD mit einem Beighton-Score-Fragebogen und einem validierten fünfteiligen Fragebogen.1,41,43 |
C |
Krankheitsorientierte Ergebnisse und Expertenmeinungen |
|
Eine frühzeitige multidisziplinäre Behandlung wie die |
C |
Expertenmeinungen und mehrere Fallstudien |
A = konsistente, patientenorientierte Evidenz von guter Qualität; B = inkonsistente oder patientenorientierte Evidenz von begrenzter Qualität; C = Konsens, krankheitsorientierte Evidenz, übliche Praxis, Expertenmeinungen oder Fallserien.
*Informationen über das SORT-Evidenzbewertungssystem finden Sie unter https://www.aafp.org/afpsort
Qualitative Studien beschreiben die Erfahrungen von Betroffenen mit Schmerzen und Behinderungen bei hEDS, die in ihren sozialen Kreisen und sogar in medizinischen Einrichtungen häufig bagatellisiert oder abgewertet werden. Da Betroffene als „normal“ angesehen werden oder die Erkrankung nicht erkannt wird.53-55
Die Komplexität des hEDS bzw. der HSD und die mangelnde Bekanntheit in der Ärzteschaft können dazu führen, dass sie die Erfahrungen der Betroffenen mit ihren EDS-Erkrankungen ignorieren oder ihnen skeptisch gegenüberstehen, welche sich möglicherweise nachhaltig negativ auf die Betroffenen auswirken kann.56 Qualitative Daten belegen nachdrücklich, dass eine frühzeitige Diagnose und eine einfühlsame, sachkundige sowie klinische Betreuung von Betroffenen sehr hilfreich wäre.53-55 Jedoch bedarf es weiterer Forschung, um bestätigen zu können, dass eine frühe Diagnose und eine qualifizierte, fürsorgliche Betreuung die Zufriedenheit der Betroffenen verbessert. Die Risiken und Nutzen invasiver Tests und Verfahren müssen bei Betroffenen mit allen Formen von EDS sorgfältig abgewogen werden, da Blutungskomplikationen, unzureichendes Ansprechen auf Regional- und Lokalanästhesie und iatrogene Verletzungen häufig sind.57
Die Ärzteschaft sollte den Betroffenen mit Empathie begegnen und sich bewusst sein, dass diese multisystemischen Erkrankung zu kognitive Defizite, negative Emotionen und eingeschränkter Aktivität führen kann.37 Hierzu sind weitere Untersuchungen erforderlich. Es existieren drei kleine Studien zu einem multidisziplinären Ansatz, die physikalische, ergotherapeutische und kognitive Verhaltenstherapie umfasst. Diese haben gezeigt, dass Angst, Depressionen, Katastrophisierung und Kinesiophobie (Angst vor Schmerzen aufgrund von Bewegung) abgenommen haben und sich die körperlichen Funktion und die Selbstwirksamkeit bei den behandelten Betroffenen verbessert haben.32,58,59 Eine ähnliche multidisziplinäre Intervention, die keine kognitive Verhaltenstherapie beinhaltete, zeigte keinen Nutzen.45
Prognose
Auf der Grundlage einer großen italienischen Fallstudie wurde ein dreiphasiger natürlicher Verlauf des hEDS beschrieben.49 In dieser Studie entwickelte sich die Symptomatik bei den Betroffenen von einer generalisierten Gelenkhypermobilität, mit oder ohne Gelenkschmerzen in der Kindheit, zu muskuloskelettale Schmerzen im Bewegungsapparat, Stürzen, verschiedene Kopfschmerzarten sowie funktionellen gastrointestinalen Störungen im zweiten und dritten Lebensjahrzehnt. Im dritten bis vierten Lebensjahrzehnt entwickelten die Betroffenen eine Gelenksteifigkeit, chronische Schmerzen im Bewegungsapparat und eine Verminderung der Fatigue. Die Prognose des hEDS/HSD ist sehr variabel und lässt sich für einzelne Betroffene nur schwer vorhersagen. Bei einer Studie über eine dreijährige Beobachtungszeit, die in einem Universitätsklinikum durchgeführt wurde, ergaben sich bei Zufallsstichproben von Kindern mit einer Vorstufe eines hEDS vier Faktoren, die den Schweregrad der Erkrankung und im geringen Maße die Entwicklung von Behinderung vorhersagten: Multisystembeteiligung, Schmerzen, Fatigue und Haltungskontrolle60. Allerdings variieren die Ergebnisse der Studie. Bei Erwachsenen waren chronische Schmerzen, gastrointestinale und urologische Probleme, Fatigue, eingeschränkte Mobilität und vielfache Verletzungen am häufigsten. Das Ergebnisse ist eine verminderte Lebensqualität, die zu einer eingeschränkten Teilnahme an Aktivitäten des täglichen Lebens führt.30,53,61
Datenquellen: Suche in MEDLINE und CINAHL nach Gelenkhypermobilität (allgemein, einschließlich EDS) und epidemiolog$.mp, Risikofaktoren, (Pathogenese oder pathogenetisch oder pathogenic$ oder pathogeny).mp, klinische Präsentation, Symptome, Diagnose, diagnostischen Kriterien, klinischem Management oder Prognose, beschränkt auf Englisch und die letzten 10 Jahre mit zusätzlicher Durchsicht der Bibliografien nach relevanten Artikeln. Die American Family Physician Editors fanden keine relevanten Belege in POEMs oder der Cochrane-Datenbank. Es wurden keine einschlägigen Leitlinien des ECRI Guidelines Trust oder der U.S. Preventive Services Task Force gefunden. Suchdaten: 25. und 26. November 2019 und 28. Oktober 2020.
Die Autorenschaft dankt der medizinischen Bibliotheksbetreuung des Gundersen Health System-La Crosse Campus und den Ärztinnen Karyn Laursen und Kerry Jedele für ihre Durchsicht und Unterstützung bei der Erstellung des Manuskripts.
Die in diesem Artikel geäußerten Ansichten sind die der Autorenschaft und spiegeln nicht unbedingt die offizielle Position des Department of Defense oder der U.S. government wieder.
Die Autorenschaft
KENNETH S. YEW, MD, MPH, ist Fakultätsmitglied der Gundersen Medical Foundation Family Medicine Residency, La Crosse, Wisconsin. Außerdem ist er klinischer Assistenzprofessor im Department of Family Medicine at the Uniformed Services University of the Health Sciences, Bethesda, Md.
KARA A. KAMPS-SCHMITT, MD, ist Hausärztin an der Gundersen Clinic, Tomah, Wisconsin.
ROBYN BORGE, MD, ist stellvertretende Programmdirektorin und Gründungsmitglied der Gundersen Medical Foundation Family Medicine Residency.
Adresskorrespondenz an Kenneth S. Yew, MD, MPH, Gundersen Family Medicine Residency Clinic, 1900 South Ave., Mailstop FB2-009, La Crosse, WI 54601 (E-Mail: kenyew63@gmail.com). Nachdrucke sind bei der Autorenschaft nicht erhältlich.
Referenzen
- Malfait F, Francomano C, Byers P, et al. The 2017 International Classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
- Tinkle B, Castori M, Berglund B, et al. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): clinical description and natural history. Am J Med Genet C Semin Med Genet. 2017;175(1):48-69.
- National Center for Advancing Translational Sciences; Genetic and Rare Diseases Information Center (GARD). Ehlers-Danlos syndromes. Updated April 20, 2017. Accessed May 22, 2020. https://rarediseases. info.nih.gov/diseases/6322/ehlers-danlos-syndromes
- Castori M, Tinkle B, Levy H, et al. A framework for the classification of joint hypermobility and related conditions. Am J Med Genet C Semin Med Genet. 2017;175(1):148-157.
- The Ehlers-Danlos Society. Hypermobile Ehlers-Danlos syndrome (hEDS) vs. hypermobility spectrum disorders (HSD): what’s the difference? Accessed June 18, 2020. https://ehlers-danlos.com/wp-content/ uploads/hEDSvHSD.pdf
- The International Consortium on Ehlers-Danlos Syndromes & Related Disorders in association with the Ehlers-Danlos Society. Diagnostic criteria for hypermobile Ehlers-Danlos syndrome (hEDS). Accessed March 2, 2020. https://www.ehlers-danlos.com/wp-content/uploads/hEDS- Dx-Criteria-checklist-1.pdf
- Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
- McGillis L, Mittal N, Santa Mina D, et al. Utilization of the 2017 diagnostic criteria for hEDS by the Toronto GoodHope Ehlers-Danlos syndrome clinic: a retrospective review. Am J Med Genet A. 2020;182(3):484-492.
- Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16:207.
- Demmler JC, Atkinson MD, Reinhold EJ, et al. Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: a national electronic cohort study and case-control comparison. BMJ Open. 2019;9(11):e031365.
- Mulvey MR, Macfarlane GJ, Beasley M, et al. Modest association of joint hypermobility with disabling and limiting musculoskeletal pain: results from a large-scale general population-based survey. Arthritis Care Res (Hoboken). 2013;65(8):1325-1333.
- Barçak ÖF, Karkucak M, Çapkin E, et al. Prevalence of generalized joint hypermobility and fibromyalgia syndrome in the children population of Trabzon: a Turkish study. Turk J Phys Med Rehab. 2015;61:6-11.
- Gocentas A, Jascaniniene N, Pasek M, et al. Prevalence of generalised joint hypermobility in school-aged children from east-central European region. Folia Morphol (Warsz). 2016;75(1):48-52.
- Mikkelsson M, Salminen JJ, Kautiainen H. Joint hypermobility is not a contributing factor to musculoskeletal pain in pre-adolescents. J Rheumatol. 1996;23(11):1963-1967.
- Remvig L, Kümmel C, Kristensen JH, et al. Prevalence of generalized joint hypermobility, arthralgia and motor competence in 10-year-old school children. Int Musculoskelet Med. 2011;33(4):137-145.
- Seçkin U, Tur BS, Yilmaz O, et al. The prevalence of joint hypermobility among high school students. Rheumatol Int. 2005;25(4):260-263.
- Singh H, McKay M, Baldwin J, et al. Beighton scores and cut-offs across the lifespan: cross-sectional study of an Australian population. Rheumatology (Oxford). 2017;56(11):1857-1864.
- Rombaut L, Malfait F, De Wandele I, et al. Muscle-tendon tissue proper- ties in the hypermobility type of Ehlers-Danlos syndrome. Arthritis Care Res (Hoboken). 2012;64(5):766-772.
- Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11): 2709-2716.
- Chiarelli N, Carini G, Zoppi N, et al. Transcriptome-wide expression pro- filing in skin fibroblasts of patients with joint hypermobility syndrome/ Ehlers-Danlos syndrome hypermobility type. PLoS One. 2016;11(8): e0161347.
- Colombi M, Dordoni C, Chiarelli N, et al. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am J Med Genet C Semin Med Genet. 2015;169C(1):6-22.
- Botrus G, Baker O, Borrego E, et al. Spectrum of gastrointestinal manifestations in joint hypermobility syndromes. Am J Med Sci. 2018;355(6): 573-580.
- Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes [published correction appears in Am J Med Genet A. 2017;173(12):3241]. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
- De Wandele I, Rombaut L, Malfait F, et al. Clinical heterogeneity in patients with the hypermobility type of Ehlers-Danlos syndrome. Res Dev Disabil. 2013;34(3):873-881.
- Fikree A, Chelimsky G, Collins H, et al. Gastrointestinal involvement in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):181-187.
- Hugon-Rodin J, Lebègue G, Becourt S, et al. Gynecologic symptoms and the influence on reproductive life in 386 women with hypermobility type Ehlers-Danlos syndrome: a cohort study. Orphanet J Rare Dis. 2016;11(1):124.
- De Wandele I, Rombaut L, De Backer T, et al. Orthostatic intolerance and fatigue in the hypermobility type of Ehlers-Danlos syndrome. Rheumatology (Oxford). 2016;55(8):1412-1420.
- Hakim A, O’Callaghan C, De Wandele I, et al. Cardiovascular autonomic dysfunction in Ehlers-Danlos syndrome—Hypermobile type. Am J Med Genet C Semin Med Genet. 2017; 175(1): 168-174.
- Atzinger CL, Meyer RA, Khoury PR, et al. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers-Danlos syndrome. J Pediatr. 2011; 158(5): 826-830.e1.
- Mastoroudes H, Giarenis I, Cardozo L, et al. Lower urinary tract symptoms in women with benign joint hypermobility syndrome: a case-control study. Int Urogynecol J. 2013; 24(9): 1553-1558.
- Castori M, Dordoni C, Morlino S, et al. Spectrum of mucocutaneous manifestations in 277 patients with joint hypermobility syndrome/ Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet C Semin Med Genet. 2015; 169C(1): 43-53.
- Zhou Z, Rewari A, Shanthanna H. Management of chronic pain in Ehlers-Danlos syndrome: two case reports and a review of literature. Medicine (Baltimore). 2018; 97(45): e13115.
- Albayrak İ, Yilmaz H, Akkurt HE, et al. Is pain the only symptom in patients with benign joint hypermobility syndrome? Clin Rheumatol. 2015; 34(9): 1613-1619.
- Grahame R. Joint hypermobility syndrome pain. Curr Pain Headache Rep. 2009; 13(6): 427-433.
- Rombaut L, Scheper M, De Wandele I, et al. Chronic pain in patients with the hypermobility type of Ehlers-Danlos syndrome: evidence for generalized hyperalgesia. Clin Rheumatol. 2015; 34(6): 1121-1129.
- Gharbiya M, Moramarco A, Castori M, et al. Ocular features in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type: a clinical and in vivo confocal microscopy study. Am J Ophthalmol. 2012; 154(3): 593-600.e1.
- Baeza-Velasco C, Bulbena A, Polanco-Carrasco R, et al. Cognitive, emotional, and behavioral considerations for chronic pain management in the Ehlers-Danlos syndrome hypermobility-type: a narrative review. Disabil Rehabil. 2019; 41(9): 1110-1118.
- Hakim A, De Wandele I, O’Callaghan C, et al. Chronic fatigue in Ehlers-Danlos syndrome—Hypermobile type. Am J Med Genet C Semin Med Genet. 2017; 175(1): 175-180.
- Grahame R. Hypermobility: overmedicalized? A debate. First opposition: I10. In: Williams D. Reproductive issues in rheumatology: do you know how to advise your patients? Rheumatology. 2012; 51(suppl 3): iii1-iii6.
- Castori M, Hakim A. Contemporary approach to joint hypermobility and related disorders. Curr Opin Pediatr. 2017; 29(6): 640-649.
- Juul-Kristensen B, Schmedling K, Rombaut L, et al. Measurement properties of clinical assessment methods for classifying generalized joint hypermobility—a systematic review. Am J Med Genet C Semin Med Genet. 2017; 175(1): 116-147.
- Urban Z; National Organization for Rare Disorders (NORD). Cutis laxa. Updated 2014. Accessed March 3, 2020. https:// rarediseases.org/ rare-diseases/cutis-laxa/
- Smits-Engelsman B, Klerks M, Kirby A. Beighton score: a valid measure for generalized hypermobility in children. J Pediatr. 2011; 158(1): 119-123, 123.e1-123.e4.
- Beighton P, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis. 1973; 32(5): 413-418.
- Bale P, Easton V, Bacon H, et al. The effectiveness of a multidisciplinary intervention strategy for the treatment of symptomatic joint hypermobility in childhood: a randomised, single Centre parallel group trial (The Bendy Study). Pediatr Rheumatol Online J. 2019; 17(1): 2.
- Engelbert RHH, Juul-Kristensen B, Pacey V, et al. The evidence-based rationale for physical therapy treatment of children, adolescents, and adults diagnosed with joint hypermobility syndrome/hypermobile Ehlers Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017; 175(1): 158-167.
- Russek LN, Stott P, Simmonds J. Recognizing and effectively managing hypermobility-related conditions. Phys Ther. 2019; 99(9): 1189-1200.
- Smith TO, Bacon H, Jerman E, et al. Physiotherapy and occupational therapy interventions for people with benign joint hypermobility syndrome: a systematic review of clinical trials. Disabil Rehabil. 2014; 36(10): 797-803.
- Castori M, Morlino S, Celletti C, et al. Re-writing the natural history of pain and related symptoms in the joint hypermobility syndrome/ Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A. 2013; 161A(12): 2989-3004.
- Tinkle BT, Levy HP. Symptomatic joint hypermobility: the hypermobile type of Ehlers-Danlos syndrome and the hypermobility spectrum disorders. Med Clin North Am. 2019; 103(6): 1021-1033.
- Russek LN, LaShomb EA, Ware AM, et al. United States physical therapists’ knowledge about joint hypermobility syndrome compared with fibromyalgia and rheumatoid arthritis. Physiother Res Int. 2016; 21(1): 22-35.
- Huston P, McFarlane B. Health benefits of tai chi: what is the evidence? Can Fam Physician. 2016; 62(11): 881-890.
- Bennett SE, Walsh N, Moss T, et al. Understanding the psychosocial impact of joint hypermobility syndrome and Ehlers-Danlos syndrome hypermobility type: a qualitative interview study. Disabil Rehabil. 2019; 1-10.
- Palmer S, Bridgeman K, Di Pierro I, et al. The views of people with joint hypermobility syndrome on its impact, management and the use of patient-reported outcome measures. A thematic analysis of open-ended questionnaire responses. Musculoskeletal Care. 2019; 17(2): 183-193.
- Terry RH, Palmer ST, Rimes KA, et al. Living with joint hypermobility syndrome: patient experiences of diagnosis, referral and self-care. Fam Pract. 2015; 32(3): 354-358.
- Berglund B, Mattiasson AC, Randers I. Dignity not fully upheld when seeking health care: experiences expressed by individuals suffering from Ehlers-Danlos syndrome. Disabil Rehabil. 2010; 32(1): 1-7.
- Wiesmann T, Castori M, Malfait F, et al. Recommendations for anesthesia and perioperative management in patients with Ehlers-Danlos syndrome(s). Orphanet J Rare Dis. 2014; 9: 109.
- Bathen T, Hångmann AB, Hoff M, et al. Multidisciplinary treatment of disability in Ehlers-Danlos syndrome hypermobility type/hypermobility syndrome: a pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013; 161A(12): 3005-3011.
- Rahman A, Daniel C, Grahame R. Efficacy of an out-patient pain management programme for people with joint hypermobility syndrome. Clin Rheumatol. 2014; 33(11): 1665-1669.
- Scheper MC, Nicholson LL, Adams RD, et al. The natural history of children with joint hypermobility syndrome and Ehlers-Danlos hypermobility type: a longitudinal cohort study. Rheumatology (Oxford). 2017; 56(12): 2073-2083.
- Pacey V, Tofts L, Adams RD, et al. Quality of life prediction in children with joint hypermobility syndrome. J Paediatr Child Health. 2015; 51(7): 689-695.
eTABELLE A
Ressourcen zu Hypermobilitätssyndromen
|
The Ehlers-Danlos Society Diagnosekriterien für das hypermobile Ehlers-Danlos-Syndrom (ausfüllbares PDF) https://www.ehlers-danlos.com/wp-content/uploads/hEDS-Dx-Criteria-checklist-1-Fillable-form.pdf |
|
Hypermobility Syndromes Association Hypermobilitäts-Störungen: Ein Update für Ärztinnen und Ärzte |
|
Royal Collage of General Practitioners Die Ehlers-Danlos-Syndrome-Toolkit (praktische Informationen) https://www.rcgp.org.uk/clinical-and-research/resources/toolkits/ehlers-danlos-syndromes-toolkit.aspx |
Wir bedanken uns ganz herzlich bei Herrn Dr. Kenneth S. Yew für die freundliche Genehmigung, seinen Artikel übersetzen zu dürfen. Unser Dank geht gleichermaßen an die Stiftung American Academy of Family Physicians (AAFP) https://www.aafpfoundation.org/home.html, die uns die Genehmigung erteilt hat, die Übersetzung online stellen zu dürfen.
