Komorbiditäten
Begleiterkrankungen

Bei den Ehlers-Danlos-Syndromen gibt eine Vielzahl von möglichen Begleiterkrankungen.
Einige dieser Begleiterkrankungen werden gehäuft zusammen beobachtet, auch wenn die wissenschaftlichen Belege zum Teil noch erforscht werden müssen. Die nachfolgende Auflistung möglicher Begleiterkrankungen hat nicht den Anspruch auf Vollständigkeit.
• ADHS/ADS
• Arthrose
• Augenerkrankungen (z.B. Netzhautablösung, hohe Myopie, Schielen, seltener Keratokonus)
• Autismus-Spektrum-Störungen (ASS)
• Beckenbodendysfunktion (PFD)
• Chiari-Malformation Typ 1
• Chronisches Fatigue Syndrom (ME/CFS)
• Craniomandibuläre Dysfunktion (CMD)
• Dysautonomien (Fehlregulation des vegetativen Nervensystems)
• Endometriose
• Fibromyalgie-Syndrom
• Gastrointestinale Erkrankungen (z.B. Reizdarm, Gastroparese, SIBO)
Ursachen, Diagnostik und Therapie nach Leitlinien
Was ist eine Gastroparese?
Die Gastroparese ist eine chronische Motilitätsstörung des Magens, bei der sich die Magenentleerung verzögert, ohne dass eine mechanische Blockade vorliegt. Die Nahrung verbleibt länger im Magen als physiologisch vorgesehen.
Typische Symptome sind:
- Übelkeit
- Erbrechen (teils von unverdauter Nahrung)
- Völlegefühl
- frühes Sättigungsgefühl
- Oberbauchbeschwerden
- Appetitverlust und Gewichtsveränderungen
Die Erkrankung kann die Lebensqualität erheblich einschränken.
Ursachen der Gastroparese
Die häufigsten Formen sind:
- Diabetische Gastroparese
- Idiopathische Gastroparese (ohne erkennbare Ursache)
- Postoperative oder postinfektiöse Formen
- Neurologische Erkrankungen
- Andere Erkrankungen (z.B. Bindegewebserkrankungen)
- Medikamentös bedingte Motilitätsstörungen
Internationale Leitlinien, unter anderem des American College of Gastroenterology (ACG) und der American Gastroenterological Association (AGA), betonen die sorgfältige Differenzialdiagnostik vor Diagnosestellung.
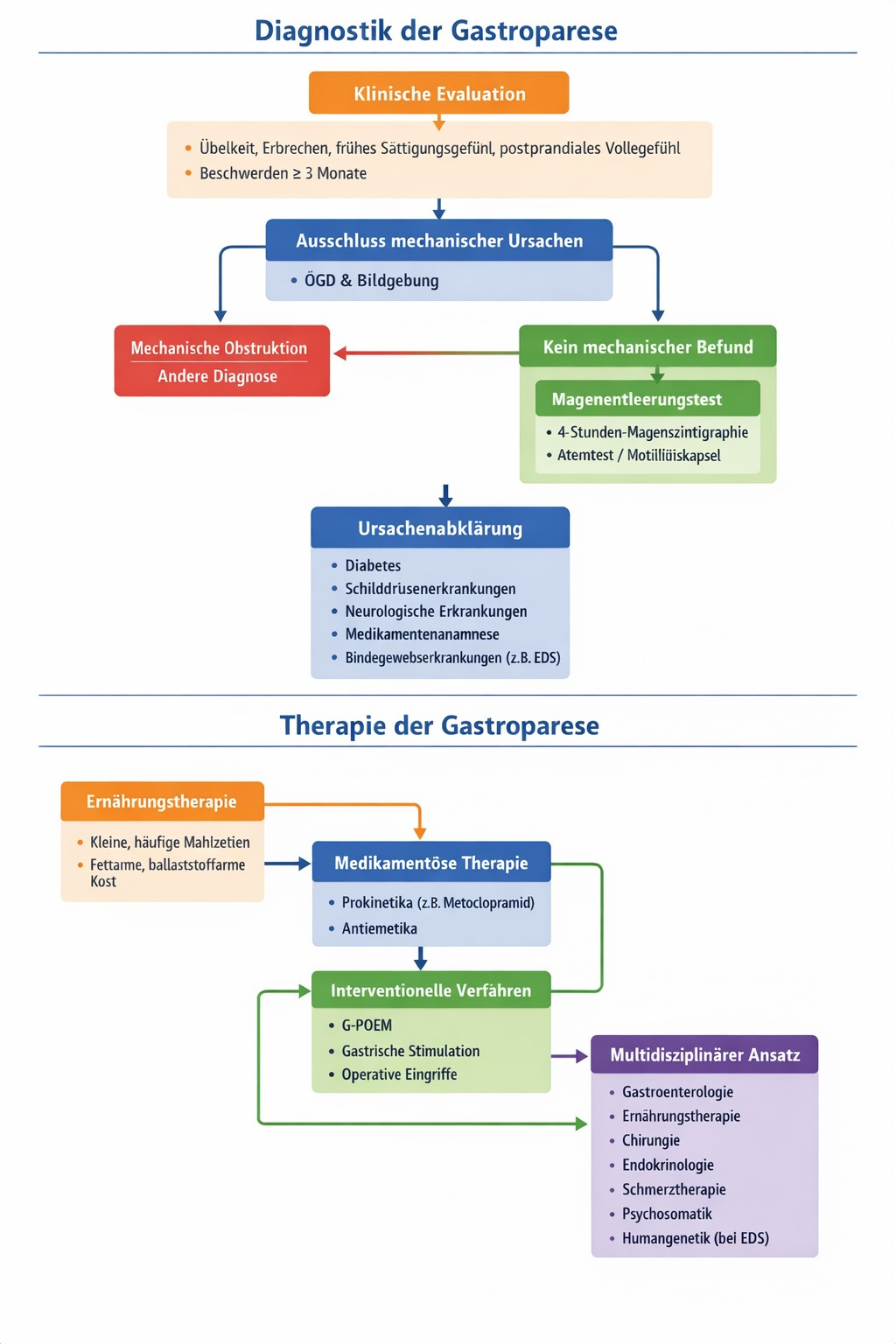
Diagnostik der Gastroparese (nach Leitlinien)
Die Diagnostik folgt klaren Kriterien, u. a. gemäß ACG- und AGA-Leitlinien.
1. Klinische Evaluation
Erfassung typischer Symptome:
- Übelkeit
- Erbrechen
- frühes Sättigungsgefühl
- postprandiales Völlegefühl
Dauer der Beschwerden: in der Regel ≥ 3 Monate.
2. Ausschluss mechanischer Ursachen
Vor Diagnosestellung muss eine mechanische Obstruktion ausgeschlossen werden, meist mittels:
- Ösophago-Gastro-Duodenoskopie (ÖGD)
- bildgebender Verfahren
3. Objektiver Nachweis der verzögerten Magenentleerung
Goldstandard ist die:
4-Stunden-Magenszintigrafie
Dabei wird die Entleerung einer standardisierten Testmahlzeit über vier Stunden gemessen. Alternativ können in spezialisierten Zentren Atemtests oder drahtlose Motilitätskapseln eingesetzt werden.
4. Ursachenabklärung
- Diabetes mellitus
- Schilddrüsenerkrankungen
- neurologische Erkrankungen
- Medikamentenanamnese
Bei klinischem Verdacht: Abklärung auf Bindegewebserkrankungen wie EDS
Eine genetische Diagnostik erfolgt nur bei entsprechender klinischer Indikation.
Therapie der Gastroparese (nach Leitlinien)
Die Behandlung erfolgt stufenweise und individualisiert.
1. Ernährungstherapie (Basis jeder Behandlung)
- Kleine, häufige Mahlzeiten
- Fett- und ballaststoffreduzierte Kost
- Gut kaubare oder pürierte Speisen
- Bei schwerer Ausprägung: Trinknahrung oder enterale Ernährung
Eine ernährungstherapeutische Begleitung ist empfohlen.
2. Medikamentöse Therapie
- Prokinetika (fördern die Magenentleerung)
- Metoclopramid (kurzfristig)
- Erythromycin (kurzfristig)
Metoclopramid ist derzeit das einzige in vielen Leitlinien empfohlene Medikament mit Zulassung für diese Indikation (unter Beachtung möglicher Nebenwirkungen).
- Antiemetika
Zur Kontrolle von Übelkeit und Erbrechen.
Die Therapie sollte regelmäßig auf Wirksamkeit und Nebenwirkungen überprüft werden.
3. Interventionelle Verfahren (bei therapierefraktären Verläufen)
Nur bei schwerer, persistierender Symptomatik:
- G-POEM (gastrische perorale endoskopische Myotomie)
- Gastrische elektrische Stimulation
- In Einzelfällen operative Verfahren
Diese Maßnahmen erfolgen in spezialisierten Zentren.
4. Multidisziplinärer Ansatz
Empfohlen wird eine interdisziplinäre Betreuung mit:
- Gastroenterologie
- Ernährungstherapie
- Chirurgie
- Endokrinologie
- Schmerztherapie
- Psychosomatik
- bei EDS: humangenetische und rheumatologische Mitbetreuung
Ziel ist nicht nur die Verbesserung der Magenentleerung, sondern vor allem die Symptomkontrolle und Steigerung der Lebensqualität.
Warum ist der Zusammenhang mit EDS klinisch relevant?
Die Erkenntnisse aus der Studie von Smieszek et al. legen nahe, dass Gastroparese bei manchen Betroffenen Teil eines systemischen Bindegewebs- oder Dysautonomie-Spektrums sein könnte.
Das hat praktische Konsequenzen:
- Sensibilisierung für Begleiterkrankungen
- frühere Diagnosestellung
- individuell angepasste Therapie
- bessere interdisziplinäre Versorgung
Gerade bei idiopathischer Gastroparese und zusätzlichen Hinweisen auf Hypermobilität oder Bindegewebsschwäche sollte an ein mögliches EDS gedacht werden.
Zusammenfassung
Die Gastroparese ist eine komplexe Motilitätsstörung mit unterschiedlichen Ursachen.
Die Diagnostik erfordert den objektiven Nachweis einer verzögerten Magenentleerung bei Ausschluss mechanischer Ursachen.
Die Therapie erfolgt leitliniengerecht stufenweise, von Ernährungstherapie über medikamentöse Behandlung bis hin zu interventionellen Verfahren.
Forschungsergebnisse deuten darauf hin, dass genetische und bindegewebsassoziierte Faktoren bei einem Teil der Patientinnen und Patienten insbesondere im Kontext der Ehlers-Danlos-Syndrome eine Rolle spielen könnten.
Leitlinien:
American Gastroenterological Association (AGA), Clinical Guideline on the Management of Gastroparesis, 2025. Link
American College of Gastroenterology (ACG), Clinical Guideline: Gastroparesis, 2022. Link
Europäischer Konsens (UEG & ESNM), Gastroparese: Empfehlungen für Diagnostik & Therapie. Link
• Gefäßkompressionssyndrome (z.B. Thoracic-outlet-Syndrom, Nussknacker-Syndrom, May-Thurner-Syndrom)
• Idiopathische intrakranielle Hypertension (IIH)
• Komplexes regionales Schmerzsyndrom (CRPS)
• Kopfgelenksinstabilität (CCI, AAI)
• Mastzellaktivierungssyndrom (MCAS)
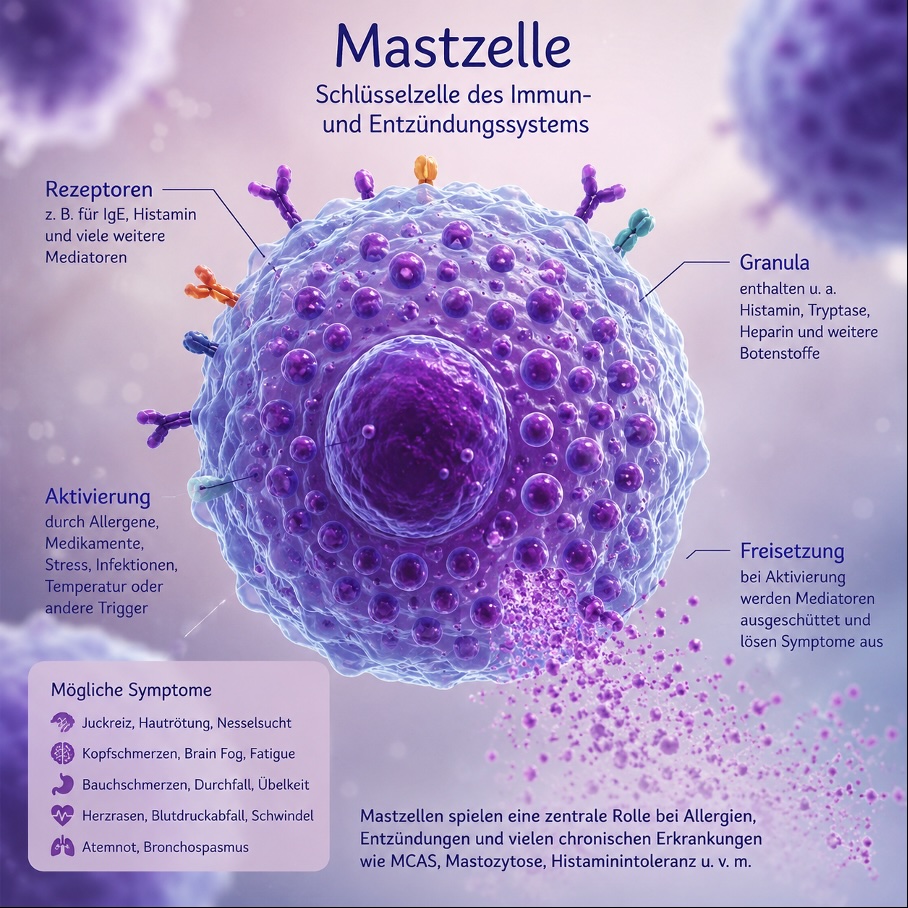
Was sind Mastzellen?
Mastzellen sind spezialisierte Immunzellen, die sich nahezu in allen Geweben des Körpers befinden. Sie spielen eine wichtige Rolle bei der Abwehr von Krankheitserregern, bei allergischen Reaktionen sowie bei der Regulation von Entzündungsprozessen. Werden Mastzellen aktiviert, setzen sie zahlreiche Botenstoffe frei, darunter Histamin, Tryptase, Prostaglandine, Leukotriene und Heparin.
Diese Botenstoffe können vielfältige Beschwerden auslösen und nahezu jedes Organsystem beeinflussen.
Was ist ein Mastzellaktivierungssyndrom (MCAS)?
Das Mastzellaktivierungssyndrom (MCAS) ist eine Erkrankung, bei der Mastzellen übermäßig oder fehlreguliert aktiviert werden und vermehrt Entzündungs- und Botenstoffe freisetzen.
Aktuelle wissenschaftliche Arbeiten gehen davon aus, dass MCAS deutlich häufiger vorkommt als bisher angenommen und oft unerkannt bleibt. Viele Betroffene erfüllen nicht die klassischen Kriterien einer Mastozytose. Zudem schließen normale Tryptasewerte eine Mastzellaktivierung nicht aus. Auch das Auftreten einer Anaphylaxie ist für die Diagnose nicht zwingend erforderlich. Die internationale Expertengruppe um Lawrence B. Afrin und Gerhard J. Molderings sieht die Unterdiagnose derzeit als größeres Problem an als eine Überdiagnose.
Typische Symptome
MCAS ist eine Multisystemerkrankung. Die Beschwerden können in unterschiedlicher Ausprägung und Kombination auftreten:
- Haut: Flush, Juckreiz, Nesselsucht (Urtikaria), Hautrötungen
- Magen-Darm-Trakt: Bauchschmerzen, Übelkeit, Durchfall, Blähungen
- Herz-Kreislauf-System: Herzrasen, Blutdruckschwankungen, Schwindel
- Nervensystem: Kopfschmerzen, Konzentrationsstörungen („Brain Fog“), Müdigkeit
- Atemwege: Atemnot, Asthma-ähnliche Beschwerden
- Bewegungsapparat: Muskel- und Gelenkschmerzen, Fatigue
- Psychische Symptome: Angstzustände, innere Unruhe oder Stimmungsschwankungen
Charakteristisch ist die Beteiligung mehrerer Organsysteme gleichzeitig.
Begleiterkrankungen
In wissenschaftlichen Veröffentlichungen werden Zusammenhänge zwischen MCAS und verschiedenen weiteren Erkrankungen diskutiert. Dazu gehören unter anderem:
- Posturales orthostatisches Tachykardiesyndrom (POTS)
- Dysautonomien
- Hypermobiles Ehlers-Danlos-Syndrom (hEDS)
- Hypermobilitäts-Spektrum-Erkrankung (HSD)
- Autoimmunerkrankungen
- Long Covid
- Myalgische Enzephalomyelitis/Chronisches Fatigue-Syndrom (ME/CFS)
- Multiple Chemical Sensitivity (MCS/TILT)
Die genauen Zusammenhänge sind Gegenstand aktueller Forschung.
Wie wird MCAS diagnostiziert?
Die Diagnostik kann herausfordernd sein, da viele Mastzellmediatoren instabil sind und Laborproben korrekt gekühlt und zeitnah verarbeitet werden müssen. Zudem können die Beschwerden unspezifisch sein und sich mit anderen Erkrankungen überschneiden.
Nach den sogenannten Consensus-2-Kriterien wird die Diagnose gestellt, wenn Beschwerden durch Mastzellmediatoren in mindestens zwei Organsystemen auftreten und zusätzlich mindestens eines der folgenden Kriterien erfüllt ist:
- Nachweis erhöhter Mastzellmediatoren (z. B. Tryptase, Histamin, N-Methylhistamin, Heparin, Prostaglandin-D₂-Metaboliten oder Leukotrien E₄)
- Vermehrte Mastzellen in Gewebeproben
- Nachweis mastzellspezifischer genetischer Veränderungen
- Deutliche Besserung unter mastzellgerichteter Therapie
Die Diagnose erfolgt daher immer auf Grundlage der gesamten klinischen Situation und nicht allein anhand eines einzelnen Laborwertes.
Therapie des Mastzellaktivierungssyndroms
Derzeit gibt es keine Standardtherapie, die für alle Betroffenen gleichermaßen geeignet ist. Die Behandlung muss individuell an Symptome, Begleiterkrankungen und Verträglichkeit angepasst werden.
Häufig eingesetzte Medikamente sind:
- H1-Antihistaminika (z. B. Cetirizin, Loratadin, Fexofenadin)
- H2-Antihistaminika (z. B. Famotidin)
- Cromoglicinsäure (Cromolyn)
- Ketotifen
- Leukotrien-Rezeptorblocker (z. B. Montelukast)
- Flavonoide wie Quercetin oder Luteolin
- Niedrig dosiertes Naltrexon (LDN)
Zusätzlich erfolgt häufig eine symptomorientierte Behandlung, beispielsweise bei Asthma, Magen-Darm-Beschwerden, Herzrasen, Dysautonomie oder POTS.
Bei schweren allergischen Reaktionen oder Anaphylaxie kann ein Adrenalin-Autoinjektor erforderlich sein.
Weitere Therapieoptionen können der unten genannten Studie entnommen werden.
Was ist eine Mastozytose?
Die Mastozytose ist eine seltene Erkrankung, bei der sich Mastzellen krankhaft vermehren und in Haut, Knochenmark oder anderen Organen ansammeln.
Man unterscheidet insbesondere:
- Kutane Mastozytose (vorwiegend Hautbefall)
- Systemische Mastozytose (Beteiligung innerer Organe)
Ursächlich liegt häufig eine Veränderung im KIT-Gen vor, die zu einer unkontrollierten Vermehrung von Mastzellen führt.
Die Symptome ähneln häufig denen des MCAS, können jedoch durch die erhöhte Anzahl von Mastzellen stärker ausgeprägt sein.
Diagnostik der Mastozytose
Die Diagnostik umfasst je nach Situation:
- Bestimmung der Basaltryptase
- Blutuntersuchungen
- Nachweis von KIT-Varianten
- Knochenmarkuntersuchung
- Gewebeuntersuchungen und histologische Analysen
Die Diagnose erfolgt nach international anerkannten Kriterien durch spezialisierte Zentren.
Therapie der Mastozytose
Auch bei der Mastozytose werden zunächst Medikamente eingesetzt, die Mastzellmediatoren blockieren oder deren Freisetzung vermindern.
Bei fortgeschrittenen Formen stehen heute zusätzlich zielgerichtete Therapien zur Verfügung. Dazu gehören insbesondere sogenannte Tyrosinkinase-Inhibitoren wie:
- Avapritinib
- Midostaurin
- Imatinib
- Dasatinib
- Nilotinib
Weitere Behandlungsoptionen können Cladribin, Interferon-α oder Hydroxyurea sein.
Trigger erkennen und vermeiden
Viele Betroffene berichten über individuelle Auslöser, die Beschwerden verstärken können. Häufig genannte Trigger sind:
- Histaminreiche Lebensmittel
- Alkohol
- Bestimmte Medikamente (z. B. Opioide, NSAR oder ACE-Hemmer)
- Hormonelle Einflüsse
- Extreme Temperaturen
- Mechanischer Druck oder Reibung
- Infektionen, Stress oder körperliche Überlastung
Das Erkennen und Vermeiden persönlicher Trigger kann ein wichtiger Bestandteil der Behandlung sein.
Fazit
Mastzellaktivierungssyndrom (MCAS) und Mastozytose gehören zu den Mastzellerkrankungen und können vielfältige Beschwerden in unterschiedlichen Organsystemen verursachen. Die Diagnostik ist häufig komplex und erfordert Erfahrung mit diesen Erkrankungen. Aktuelle Studien betonen die Relevanz einer individuellen Behandlung anhand der Beschwerden, Begleiterkrankungen und Bedürfnissen von Betroffenen.
Zusammenhang zwischen hEDS/HSD, POTS und MCAS
In den vergangenen Jahren hat das Interesse an möglichen Zusammenhängen zwischen dem hypermobilen Ehlers-Danlos-Syndrom (hEDS), der Hypermobilitäts-Spektrum-Erkrankung (HSD), dem Posturalen orthostatischen Tachykardiesyndrom (POTS) und dem Mastzellaktivierungssyndrom (MCAS) deutlich zugenommen.
Viele Betroffene berichten über Symptome aus allen drei Bereichen. Dazu gehören beispielsweise Gelenkhypermobilität und chronische Schmerzen, Kreislaufbeschwerden mit Herzrasen und Schwindel sowie Beschwerden, die mit einer Mastzellaktivierung in Verbindung gebracht werden, wie Flush, Juckreiz, Magen-Darm-Beschwerden oder Unverträglichkeitsreaktionen.
Wissenschaftliche Studien zeigen, dass diese Erkrankungen häufiger gemeinsam auftreten können als in der Allgemeinbevölkerung. Die genauen Ursachen dieser Zusammenhänge sind jedoch noch nicht vollständig geklärt. Diskutiert werden gemeinsame genetische Faktoren, Veränderungen des Bindegewebes, Störungen des autonomen Nervensystems sowie Auswirkungen von Mastzellmediatoren auf Gefäße, Nerven und Bindegewebe.
Die aktuelle wissenschaftliche Literatur weist darauf hin, dass Mastzellaktivierung bei einem Teil der Betroffenen eine verstärkende Rolle spielen könnte. Gleichzeitig besteht jedoch weiterhin Forschungsbedarf, um die genauen biologischen Zusammenhänge besser zu verstehen.
Für Betroffene ist es wichtig zu wissen, dass Symptome nicht isoliert betrachtet werden sollten. Eine interdisziplinäre Diagnostik kann helfen, mögliche Begleiterkrankungen zu erkennen und individuelle Behandlungsstrategien zu entwickeln.
Quellenangaben:
Afrin, L., Blitshteyn, S., Bluestein, L., Dempsey, T., Maxwell, A., Miller, C., Nagy, È., Nugent, D., Schofield, J., Weinstock, L., Xi, S. & Molderings, G. (). Progress in mast cell activation syndrome: the global consensus-2 diagnostic criteria at six years. Diagnosis. https://doi.org/10.1515/dx-2026-0016